![]()
scPagwas employing the polygenic regression model to uncover trait-relevant cell subpopulations by incorporating pathway activity transformed scRNA-seq data with genome-wide association studies (GWAS) data.
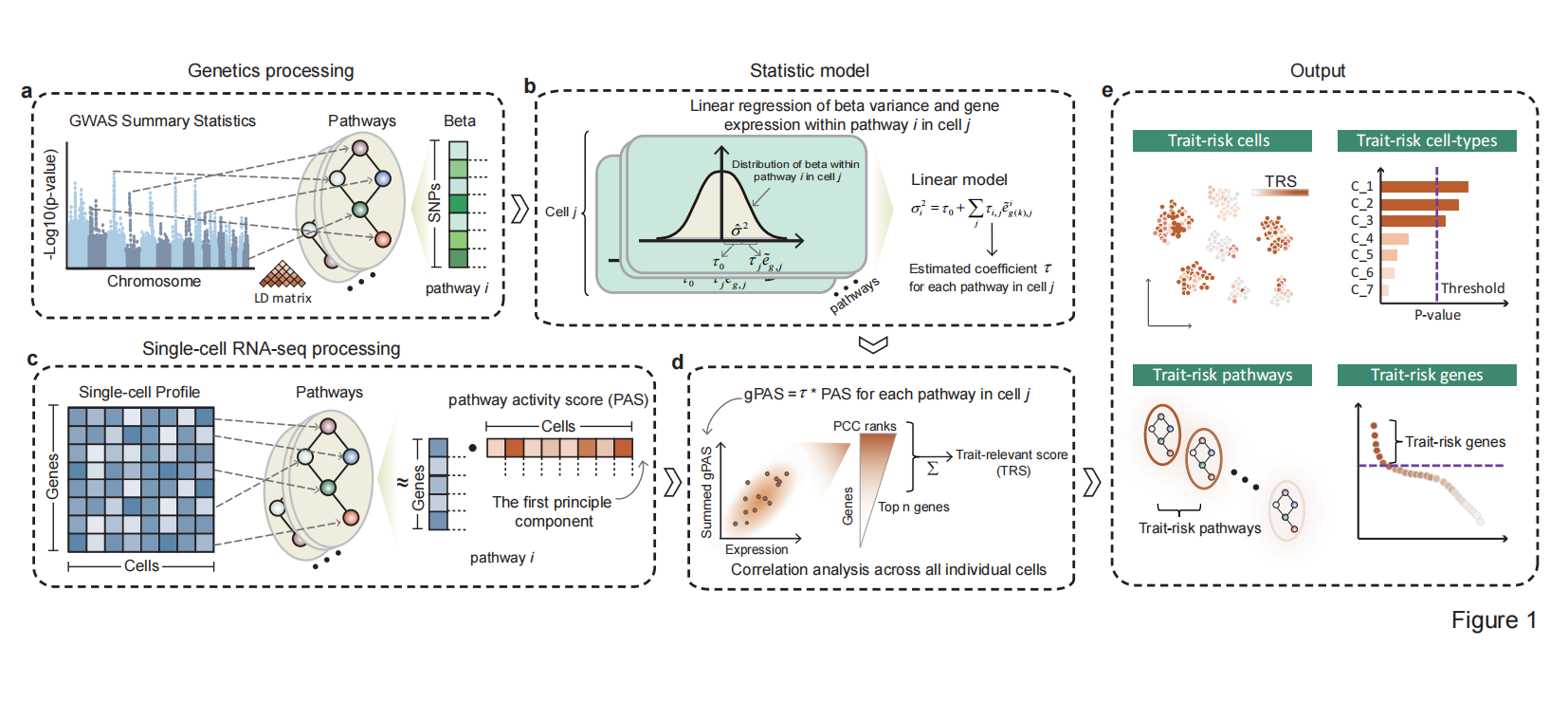
Please cite this article in press as:Ma et al.,Polygenic regression uncovers trait-relevant cellular contexts through pathway activation transformation of single-cell RNA sequencing data,Cell Genomics (2023),https://doi.org/10.1016/j.xgen.2023.100383
Code for reproducing the analysis from the paper is available here, or ![]() You can install the released version of scPagwas from github with:
You can install the released version of scPagwas from github with:
#install some dependence packages
install.packages("Seurat")
install.packages("ggpubr")
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GenomicRanges")
BiocManager::install("IRanges")
devtools::install_github("sulab-wmu/scPagwas")
In many cases, installing packages using devtools::install_github may fail. In such situations, an alternative approach is to download the package from a provided source URL and install it locally.
We also uploaded the installation package onto the Baidu cloud disk. If you’re unable to connect to GitHub or access the internet, you can download it from here. I’ll keep it updated in sync with GitHub. Link: https://pan.baidu.com/s/1X7pRzevQUIfLIATd2I0Dvw?pwd=1234 Extract code: 1234
Usage
quick-start example:
library(scPagwas)
#1.start to run the wrapper functions for example.
Pagwas_data<-scPagwas_main(Pagwas = NULL,
gwas_data =system.file("extdata", "GWAS_summ_example.txt", package = "scPagwas"), # The GWAS Summary statistics files
Single_data =system.file("extdata", "scRNAexample.rds", package = "scPagwas"),# scRNA-seq data in seruat format with "RNA" assays and normalized.
output.prefix="test", # the prefix name for output files
output.dirs="scPagwastest_output",# the directory file's name for output
block_annotation = block_annotation,# gene position in chromosome is provided by package.
assay="RNA", # the assays for scRNA-seq data to use.
Pathway_list=Genes_by_pathway_kegg,# pathway list is provided by package, including gene symbols.
n.cores=1,
iters_singlecell = 10,
chrom_ld = chrom_ld,# The LD data is provided by package.
singlecell=T, # Whether to run the singlecell process.
celltype=T# Whether to run the celltype process.
)